Chapter 6: Personalized proteomics of human biofluids for clinical applications
Doi: 10.4155/FSEB2013.14.1
Authors
Martin Andreas Feig1*
Nico Jehmlich1,2*
Uwe Völker1
Elke Hammer#
University Medicine Greifswald
Interfaculty Institute for Genetics and Functional Genomics
Department of Functional Genomics
Friedrich-Ludwig-Jahn-Str. 15A
17475 Greifswald, Germany
Tel: +49 3834 86 5872
Fax: +49 3834 86 795872
e-mail: [email protected]
1University Medicine Greifswald, Interfaculty Institute for Genetics and Functional Genomics, Department of Functional Genomics, Germany
2Department of Proteomics, Helmholtz-Centre for Environmental Research – UFZ, Germany
*contributed equally, # corresponding author
About the Authors
Martin A. Feig
Martin A. Feig is enrolled at the Ernst-Moritz-Arndt-University Greifswald (Germany) as a Master of Science student. He completed his Bachelor’s in Biomedical Science in 2014 and earned his medical degree (MD) in late 2015. Since 2011 he is doing research at the Department of Functional Genomics at the Interfaculty Institute for Genetics and Functional Genomics in the field of human biofluid proteomics, focusing on its potential in individualized medicine and therapy outcome prediction.
Nico Jehmlich
Since April 2013 Nico Jehmlich is group leader at the Department of Proteomics for biodegradation and microbial ecology at the Helmholtz Centre for Environmental Research – UFZ, Leipzig. From 2009- 2013 he worked as postdoc at the Department of Functional Genomics at the Interfaculty Institute for Genetics and Functional Genomics supervised by Prof. Dr. Uwe Völker at the University Medicine Greifswald. His research was focused on personalized proteomics of human biofluids.
Elke Hammer
Elke Hammer is employed at the Department of Functional Genomics of the Interfaculty Institute for Genetics at the University Medicine in Greifswald. She is a biologist from profession and moved in 2004 from the field of Applied Microbiology and the analysis of small molecules to the in depth analysis of protein profiles in clinical specimens as tissue and biofluids with special focus on cardiovascular diseases. As a proteomics group leader in a core facility, published papers cover relative and absolute quantitation of proteins in bacteria, mammalian cell lines, animal and human tissues and biofluids as well as protein-protein interactions.
Uwe Völker
Uwe Völker is Head of the Department of Functional Genomics of the Interfaculty Institute of Genetics at the University Medicine at the Ernst-Moritz-Arndt-University Greifswald. In his research he is applying functional genomics technologies for the elucidation of cellular adaptation reactions to environmental cues. His main fields of interest are the study of bacterial host-pathogen interactions and cardiovascular diseases. He has contributed to more than 200 articles.
Personalized proteomics of human biofluids for clinical applications
Introduction
Development of personalized medicine approaches remains challenging, but technologies including proteomics provide novel opportunities to identify biomarker candidates to accelerate the implementation of new diagnostic tests. In this chapter, we will review the current state of the proteome catalogues of the major human biofluids of plasma, urine, liquor and saliva. Furthermore, the different levels of variabilities will be discussed. Finally, large-scale plasma proteome studies will be presented that provide promising approaches to bridge the gap from biomarker discovery towards clinical practice.
Human biofluids
Introduction to human biofluids
The term biofluid is used for those fluids that contain approximately 40% of the (human) body water. Biofluids are excreted, secreted or provided from the body and possess different characteristics and compositions, and exert numerous functions, such as transport and signaling purposes (e.g., plasma), cooling (e.g., sweat), immunoactivity (e.g., cerumen), intra-individual transport (e.g., chyle), inter-individual transport (e.g., breast milk), digestion (e.g., pancreatic juice), lubrication (e.g., peritoneal fluid), as a place holder (e.g., vitreous humor) or for detoxification (e.g., urine). Human biofluids possess a complex composition that can be used in clinical practice and some of them (e.g., plasma, urine, saliva) are even accessible without invasive procedures and physical stress for the patients (Table 1). Thus, some body fluids, such as plasma, urine and liquor, are routinely and intensively used in clinical diagnostics, whilst for others, such as saliva, clinical applications are tested and yet others have not been implemented in medicine so far. Their composition and complex changes associated with disease are not comprehensively explored, yet.
Table 1: List of human biofluids categorized by their current practice in clinical routine analysis.
| Used in clinical routine | Increasing clinical relevance |
No clinical relevance yet |
| Plasma | Bile | Aqueous humor |
| Urine | Breast milk | Cerumen |
| Liquor (CSF) | Gastric juice | Cervicovaginal fluid |
| Saliva | Menstrual blood | Chyle |
| Amniotic fluid | Nasal mucus | Chymus |
| Bronchoalveolar lavage fluid | Nipple aspirate fluid | Epithelial Lining fluid |
| Feces | Pericardial fluid | Interstitial fluid from solid tumors |
| Seminal fluid | Peritoneal fluid | Lymph |
| Sputum | Pus | Perilymph |
| Sweat | Synovial fluid | Rheum |
| Tears | Sebum | |
| Vaginal secretion | Smegma | |
| Vitreous humor | Uterine fluid | |
| Vomit |
Challenges of human biofluid proteomics
Variabilities substantially influence the results of human biofluid proteome analysis. The levels of variability can be grouped into parameters affecting: i) protein composition of body fluids in terms of abundance and time caused by, for example, gender, age or circadian rhythm (= intra- and inter-subject variability); ii) the sample composition caused by, for example, sampling, depletion, fractionation, separation or storage (= processing variability); iii) the measurability of the proteome caused by, for example, liquid chromatography (LC) conditions, mass spectrometric data acquisition or the sensitivity and accuracy of the used MS platform (= LC-MS based variability) and finally; iv) the data analysis caused by, for example, non-standardized algorithm, normalization methods, thresholds and confidence (= data analysis variability) (Figure 1). Provided that sampling, processing, measurement and data analysis follow well-established and standardized workflows, inter-subject biological variation makes by far the greatest contribution to overall variability in the generated data [1]. Therefore, the proteomic study design is of utmost importance for a successful body fluid investigation to reveal, for example, disease-specific marker candidates. Phenotype characteristics need to be precisely defined and measurements acquired according to high-quality standards. Thus, a clinical routine analysis is needed and that calls for carefully designed clinical trials or well-controlled population-based approaches.
The extreme dynamic range of protein abundances, which often spans more than ten orders of magnitude, is a particular challenge associated with biofluid analysis [2]. Thus, for example in plasma, low pg/ml for interleukins and tissue leakage proteins have to be determined against a background of high abundant plasma proteins that are present in 3 × 1010 pg/ml. Global protein profiling of this huge abundance is still challenging owing to a high protein content of 60–80 mg/ml and a dynamic range of 9–12 orders of magnitude. Only 22 proteins account for 99% of the total protein content [3]. Few proteins dominate the total protein pattern and therefore obscure the detection of proteins present at much lower concentrations in MS-based approaches. To assess the remaining 1%, a wide variety of methods for depletion of the abundant proteins and gel-based and gel-free pre-fractionation have been developed. Therefore, even for in-depth characterized proteomes, many proteins are not covered in standard proteomics [2]. Anderson et al. showed that proteins used as clinical biomarkers have only been detected by HUPO’s Plasma Proteome Project in the higher concentration ranges of mg/ml and µg/ml levels [4].
A large dynamic range of abundances is characteristic for all biofluids, but the extent differs. Experimental procedures have to take this particular challenge into account. Potential approaches would be depletion of abundant proteins or approaches for extensive fractionation. However, depletion of abundant proteins removes many other proteins as well, generating a particular remaining sample, the depletome [5]. Extensive fractionation on the other side is enormously time-consuming and not compatible with the analysis of large sample sets that are required for coverage of statistically significantly associated and meaningful changes against the background of the inter-individual variation. Alternatively, targeted analysis of pre-selected candidates by immunoaffinity capture can dramatically enhance the sensitivity of measurements. The stable isotope standards and capture by anti-peptide antibodies (SISCAPA) approach has been developed to enrich low-abundance peptides from complex mixtures [6].
Compared to single bacterial or human cell cultures, protein identification in human biofluids is remarkably low. In order to increase the protein identification rate, a comprehensive and exhaustive preparation process before LC-MS measurement is needed; however, this is almost impracticable in clinical practice. In addition, peptide analyses with LC gradients of up to 4 hours permit much lower throughput compared to microarrays or ELISA assays. However, proteomics is a promising technique that is well-suited to characterizing the current physiological status and detecting structural protein modifications.
Figure 1: Different levels of variability that influence human biofluid analysis. Intra/inter-subject variability, processing variability, LC-MS based variability, and data analysis variability.
Proteome catalogues of the major human biofluids
Since human biofluids are in direct contact with a variety of tissues and organs, they can be used to describe local pathologies as well as systemic diseases to assess an individual’s health state. Proteomics has been used to screen for biomarker candidates in a few biofluids, which were then subjected to in-depth proteome analysis, including the major fluids of plasma, urine, cerebrospinal fluid and saliva. For some unconventional biofluids, such as cerumen, first proof-of-principle studies have been performed [7] but the remaining the proteome catalogue still needs to be explored.
Human blood (and its liquid component plasma or serum) is routinely collected as clinical specimens from patients. Plasma is obtained as supernatant after centrifugation of a collection tube of fresh blood containing an anticoagulant. Blood plasma is more often used than serum since the in vitro process of coagulation to obtain serum may introduce variation by co-agglutination of proteins. Plasma contains dissolved proteins with concentrations ranging from approximately 3 × 1010 pg/ml for albumin to the low pg/ml range for some cytokines and secreted or leaking proteins. Successively, the number of proteins identified by LC-MS was increased from 100 to 1929, which is up to now the most comprehensive plasma proteome described [8].
Urine as human biofluid qualifies for disease-specific research because it can be easily, noninvasively and repeatedly collected from single subjects. Proteins present in urine are derived not only from glomerular ultrafiltration of plasma but also from tubular secretion of soluble proteins, or exosome shedding through the urothelium. For healthy individuals, 30% of the urinary proteome has been estimated to originate from the plasma filtrate whereas the remaining 70% is believed to be derived from the kidneys and the urothelium. Until 2005, 800 urinary proteins had been identified by different proteomic techniques. In 2006, a comprehensive proteomic study identified more than 1500 proteins from healthy human urine samples, simultaneously reflecting the complexity and the potential information concealed in the urinary proteome. In 2009, Kentsis et al. reported the hitherto largest data set for the urinary proteome, unveiling more than 2300 proteins [9].
Cerebrospinal fluid (CSF), often termed as liquor, is clinically used as a diagnostic fluid for diseases such as meningitis, myelitis and multiple sclerosis. CSF conducts nutrition and signal transduction where blood is excluded owing to the hematoencephalic barrier. CSF has a very low protein concentration that hinders the proteome analysis. However, determining the composition and the changes in protein abundance is important to identify diagnostically relevant protein candidates for stratification of neurological disorders. LC-MS is not a standard technique for liquor diagnostics but is now being applied more often. Pooled CSF was comprehensively analyzed using LC-MS coupled with immunoaffinity depletion and strong cation exchange chromatography methods and resulted in the identification of about 4364 proteins [10].
Human whole saliva consists of different components, including gingival crevicular fluid, expectorated bronchial and nasal secretions, serum and blood derivatives from oral wounds, bacteria, desquamated epithelial cells, other cellular components, and food debris [11]. Whole saliva is often examined in salivary-based studies. Extensively investigated were Sjögren’s syndrome, periodontitis and oral cancer, which all cause alterations of the salivary protein pattern [12]. In-depth proteomic characterization of human whole saliva was accomplished by Bandhakavi et al. to maximize the proteome coverage and catalogue 2304 salivary proteins [13].
Cerumen, as a non-major human biofluid, was examined for in-depth proteome characterization. Thus, 1D-PAGE and online SCX-fractionation coupled to LC-MS was accomplished and resulted in 2013 distinct protein identifications. It was shown that cerumen shares the typical attribute of human biofluids having a high content of high-abundance proteins, such as mucin-like protein 1, prolactin-inducible protein, apolipoprotein D and zinc-alpha-2-glycoprotein, which accounts for 80% of the relative intensity of all measured proteins. In future applications, cerumen analysis may address local infections or malignancies non-invasively.
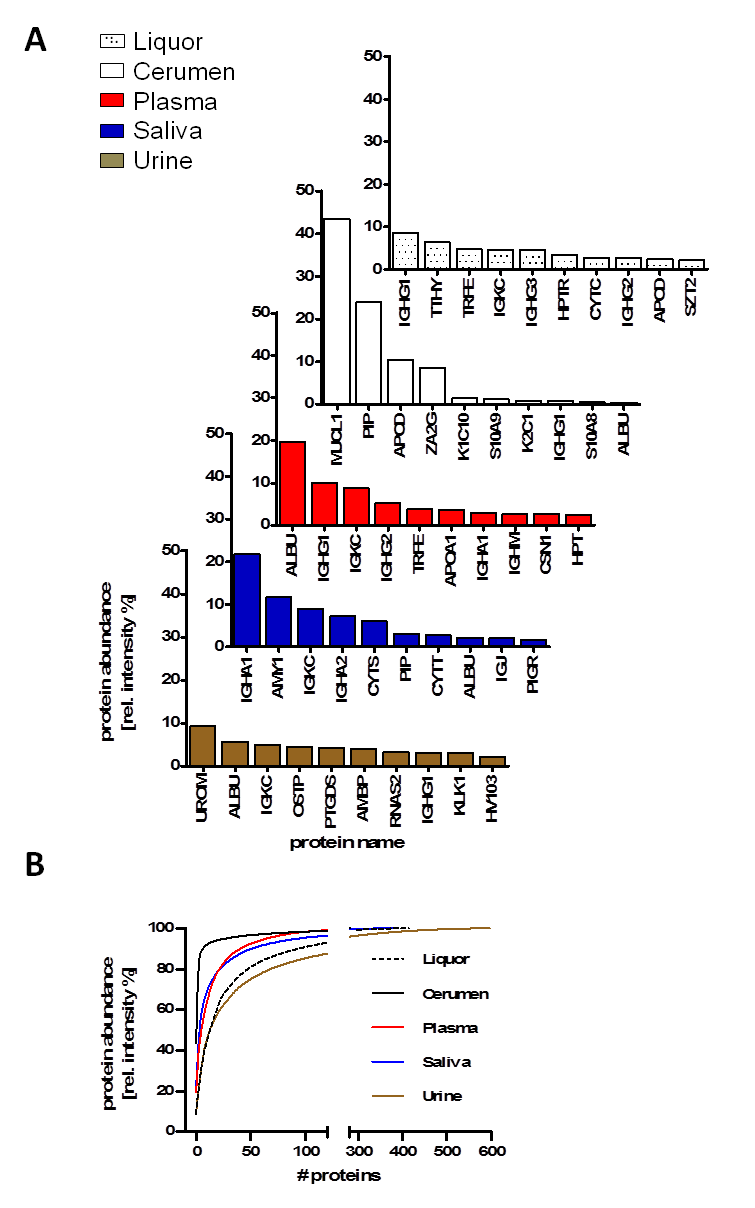
Among the “variable” proteome that contributes to their specific function, a “core” proteome of human biofluids was observed. We compared the proteome of five human biofluids to indicate the overlapping protein part (= core proteome) after standardized single-run LC-MS analysis (Figure 2). The core proteome provides an option to explore eminent pathological variance in large-scale proteome analysis of population-based cohorts where depletion and/or fractionation are too time-consuming and labor-intensive. Depth of protein identification depends on the abundance (dynamic range) of each biofluid. The presence and distribution of the 10 most abundant proteins, such as immunoglobulin chains and albumin, is shown throughout the major biofluids whereas some proteins are specific, for example, uromodulin for urine or amylase for saliva (Figure 2A). The cumulative relative protein abundance observed in single in-house LC-MS analysis for plasma, urine, cerebrospinal fluid, cerumen and saliva of the 10 most abundant proteins is plotted in Figure 2B.
Figure 2: Comparison of protein identifications and protein abundances of human biofluid proteomes observed in in-house LC-MS analysis of plasma, urine, cerebrospinal fluid, cerumen and saliva. (A) Distribution of the Top-10 protein abundance in plasma, urine, cerebrospinal fluid, cerumen and saliva. (B) Cumulative relative protein abundance as observed in single in-house LC-MS runs for plasma, urine, cerebrospinal fluid, cerumen and saliva of the 10 most abundant proteins. Adapted from [7] with permission © Elsevier (2013).
Advances in human biofluid proteomics
Technical advances in proteomics such as protein/antibody chips, depletion of high abundant proteins or equalization techniques can be accomplished by multi-affinity removal systems (MARS), affinity enrichment of targeted protein analytes or unspecific hexapeptide libraries as well as multidimensional chromatographic fractionation. All of these techniques focus on the detection of proteins in biofluids across a broader dynamic range and make the detection of disease-relevant proteins in fluid biospecimen possible [14,15]. In addition, multi-step depletions or 1D up to 4D protein/peptide separation can indeed improve the protein identification rate. However, up to ten-fold more starting material is needed. Further progress can be achieved by mass spectrometry-based improvements accomplished by higher scanning speed and sensitivity coupled with robust and reproducible measurement over long time and large samples sets [16].
Two common strategies to discover diagnostic or prognostic biomarker candidates are used: (i) comparative proteomics of a small/medium (n = 5–30) set of patients versus control samples and (ii) large-scale proteomics of population-based or clinical cohorts. Results from case-control experiments (i) enable conclusions to be drawn from associations between protein abundances and modifications and the disease states regarding their onset, progression or therapeutic monitoring. Post-translational modifications, including phosphorylation, glycosylation, acetylation or oxidation, are of great interest as they have been demonstrated to be linked to disease pathology. In addition, this strategy can provide a profound view on the proteome dynamics but is limited in the statistics owing to low number of samples analyzed. Therefore, the number of false positive hits might be higher and more extensive validation experiments have to be performed. In contrast, clinical and population-based cohorts (ii) provide, besides a standardized sampling of biospecimens, a comprehensive medical patient characterization, too. This helps to reduce the influence of sampling variation, which together with a stable and reliable liquid chromatography/MS system is a prerequisite for the detection of changes attributable to the biological state rather than to the technical variability that can occur by screening cohorts of up to several hundred samples. Such large-scale analysis offers new possibilities to compute advanced correlation analyses on proteome levels.
A critical challenge is the lack of an ability to accurately and reproducibly measure a meaningful number of proteins across institutions, mostly owing to the missing standardized MS instrumentation platform and the high variability of human biofluids. Therefore, the intra- and inter-individual variances of the fluid have to be determined before cut-off values for biomarker candidates are defined. A proper experimental study design including selection and definition of the appropriate control and/or healthy group is necessary.
Status of human biofluid proteomics and its impact on biomarker development
Highly relevant biofluids, such as plasma, urine, CSF or saliva, have been characterized by proteomic studies because of high expectations to identify promising candidates. Further biofluids with minor clinical relevance but with a diagnostically relevant potential were recently characterized such as cerumen, bile or cervicovaginal fluid. The recent status of in-depth proteome studies of the most prevalent human biofluids is indicated in Table 2 [7-9,13,17-20]. In total, 8276 protein groups were identified across all nine considered proteome studies. Interestingly, about 159 proteins overlap in all nine proteome studies of human biofluids, which means that about 4–16% of the proteins can be defined as ubiquitously occurring in biofluid proteomes. About 4 to 44% of the proteome remains unique to each fluid so that diagnostically relevant markers might be identified in this group. However, a major proportion of the proteome overlaps with one or more other biofluids and therefore disease-specific information might be redundantly represented by biofluids. In 2012, Higashi reported that non-protein hormone levels (17(OH)-progesterone) measured by LC-MS correlates well in saliva and blood patient samples and therefore both biofluids can be used to monitor cortisol administration [21]. Painful patient cannulation obtaining plasma might be substituted by non-invasive saliva collection. Notably, the protein overlap between plasma and saliva is about 50%, and the gene ontology distribution showed in all major categories high similarities [22]. Salivary proteins were only over-represented in catalytic function, protein metabolic and catabolic processes, which indicates the major function of saliva in food lubrication and first digestion.
The overall goal for clinical proteomic investigation of disease-relevant changes is bridging the gap from discovery to clinical implementation. Typically, reported significant protein candidates include well-known and abundant proteins, such as albumin, subsets of immunoglobulins, apolipoproteins, alpha-2-macroglobulin, and ceruloplasmin or complement factors. However, detected effects often reflect only the immune response and do not mirror the disease status [23]. Promising candidate proteins, such as zinc-alpha-2-glycoprotein, vary in protein abundance in the presence of disease states, including acute kidney injury [24], prostate [25], liver or breast cancer [26]. However, no direct pathophysiological correlation has been disclosed so far. A successful story is the proteome identification of five protein biomarker candidates by SELDI-TOF MS in a multi-institutional analysis including more than 600 individuals in order to discriminate ovarian cancer. In 2009, the US FDA approved the OVA1-test combined with the already established detection of CA125 in order to assess the malignancy of ovarian cancer as a method of triaging women with masses [27].
A critical component to meet FDA requirement for analytical validation of protein biomarker was intensively discussed by Boja et al. [14] regarding the evaluation and clinical usage of MS platforms for the analyses of biomarker candidates by a targeted multi-reaction monitoring (MRM) approach. High-resolution mass spectrometers and liquid chromatography systems from different manufacturers resulted in different and non-equivalent performance of the specific diagnostic assays. Therefore, a separate specification for each MRM platform is required for the clinical assay, which should also include internal standards. However, the immense effort complicates further diagnostic assay development. In addition, reproducible MS data across different laboratories and/or countries need to fit into the FDA requirements to accept protein biomarkers for clinical routine. If an analytical MS platform successfully demonstrated a biomarker discovery, it would be necessary to equivalently transfer it to different MS platforms, including separate evaluations of the clinical assay.
Protein candidates identified through comparative proteomics of small patient groups (n = 5–10) often failed to reproduce in independent case-control studies. In reality, effect sizes are small and therefore the diagnostic index is low to reliably reproduce in clinical diagnostics. In general, only a very small proportion of the proteome is accessible by standardized tests (0.5–1%), thus identified effects may not be transferable into effective diagnostic tests [4]. Therefore, the findings of disease-associated alterations as well as the specificity and sensitivity of the alteration need to be verified in independent population-based cohort studies. Many research groups are interested to implement accurate, efficient and reproducible protein assays (diagnostic tests) that could drive high value to the clinics. To achieve this goal, translational collaboration and communication of research groups and clinicians is of fundamental importance in order to use standardized technological platforms, standard operating procedures (SOP), good laboratory practice (GLP), data analysis standards and open access proteome databases [28]. Cloud databases have the potential to enable national and/or international community efforts to interpret the results integrationally; meta-data collections are performed by HUPO, Sys-BodyFluid, Human Protein Atlas Project, PeptideAtlas, MAPU or the Human Proteinpedia, but a centralized, open-access data repository of proteomics studies is still missing.
Table 2: Comparison of human biofluid in-depth characterization proteomic studies.
| Human biofluid | Identified proteins |
Non-redundant matched protein IDs |
Unique ID`s to biofluid/study |
Reference | MS platform | Putative clinical relevance |
| Plasma | 1929 | 1742 | 174 (10.0%) | Farrah et al. (2011) | multiple platforms, collected data set | Infection, myocardial infarction, liver failure, renal failure, prenatal test, newborn testing |
| Urine | 3435 | 3030 | 1000 (33.0%) | Kentsis et al. (2009) | SDS-PAGE, HPLC, LTQ-Orbitrap | Urinary tract infections, urothelial carcinoma, renal carcinoma, renal failure, cholangiocarcinoma |
| Cerebrospinal fluid | 4364 | 3660 | 1594 (43.6%) | Schutzer et al. (2011) | Immunoaffinity, SCX, LTQ-Orbitrap Velos | Neurodegenerative diseases, Alzheimer’s or Parkinson’s dementia, ALS, Huntington’s disease |
| Saliva | 2304 | 2048 | 283 (13.8%) | Bandhakavi et al. (2009) | Hexapeptide library, SCX, OFFGEL, LTQ-Orbitrap XL | Parodontosis, Sjögren’s disease, caries, mucosal cancers, mucositis, oral cancer squamous cell carcinoma |
| Cerumen | 2013 | 1955 | 223 (11.4%) | Feig et al. (2013) | SDS-PAGE, 2D-Salt Plug, Q Exactive | Acute/chronic otitis externa, Infection specific pattern, malignancies in outer ear canal, breast cancer |
| Bile | 2552 | 2387 | 443 (18.6%) | Barbhuiya et al. (2011) | SDS-PAGE, SCX, OFFGEL, LTQ-Orbitrap Velos | Primary biliary cirrhosis and primary sclerosing cholangitis, biliary stones, cholangiocarcinoma |
| Seminal fluid | 2545 | 2313 | 403 (17.4%) | Rolland et al. (2013) | multiple platforms, collected data set | Sperm maturation, orchitis, epididymitis, prostate cancer, testicular cancer, fertility testing, |
| Tear fluid | 1543 | 1306 | 58 (4.4%) | Zhou et al. (2012) | Offline SCX, Triple TOF 5600 | Dry eye syndrome, Basedow’s disease, Sjögren’s disease, breast cancer, antimicrobiocity, Ocular allergy |
| Cervico-vaginal fluid | 1107 | 1021 | 108 (10.6%) | Zegels et al. (2010) | SCX, SDS-PAGE, linear ion trap | Preterm birth, preeclampsia, HPV infection, HIV resistance, bacterial vaginosis, fertility testing |
Towards personalized proteomics
Personalized proteomics is the establishment of approved and accepted protein biomarker measurement aimed at individuals. For this purpose, a person’s default state has to be recorded to be able to monitor significant changes. However, without proper study design and implementation of robust analytical techniques, the effort and expectations to make the biomarker a useful reality in the near future can easily be hampered [15].
Acquisition of individual protein patterns tends towards single run LC-MS measurement instead of technical replicate measurements owing to the former development of robust and reliable platforms. Single runs are well-suited if the biofluid material is limited or the available MS time needs to be in an appropriate balance to the protein identification rate.
Proteomics based on large-scale studies has great promise to identify meaningful biomarker candidates. Zhang et al. first used LC-MS-based proteomics to discover serum peptides/proteins that varied significantly between type 1 diabetic and control subjects. Secondly, they verified these candidate peptide biomarkers using a targeted multiplexed multiple reaction monitoring (MRM) LC-MS assay in a large-scale cohort of 150 subjects and then peptide biomarkers were further validated in an independent 20-sample set blinded to the investigators [29]. Furthermore, population-based cohorts with longitudinal sampling of biofluids and personal data over decades are valuable resources. Individuals who develop a disease at a later time point can be selected and biofluid specimens collected earlier can be screened for prognostic biomarker discovery. Another promising approach to discover marker candidates is to assess the association between cis regulatory SNPs and the abundance of individual peptides, as performed by Johansson et al. These authors quantified more than a thousand peptides in each of over 1000 individuals from a population cohort with a high degree of relatedness and a known genetic structure. The association of genetic variation with abundance level of peptides was then analyzed by combining MS with genome-wide SNP data [30]. The results show that a better understanding of the impact of genetic variation on the plasma proteome is important for evaluating potential biomarker candidates for common diseases. Personalized medicine driven approaches, such as the integrative personal omics profile (iPOP), try to measure the physiological states from a single subject by multiple high-throughput methods [31]. Results from genomic, transcriptomic, proteomic, metabolomic, and autoantibody profiles measured over a long time period will uncover dynamic changes in healthy and diseased conditions. However, successful verification of new protein candidates of disease states depends primarily on the choice of methods owing to the availability of antibodies and their performance. In contrast, protein-based targeted immunoprecipitation MRM assays offer a faster, less costly approach than ELISAs and could expand the scope of targeted protein quantitation in biology and medicine [32].
Acknowledgements
There are no conflicts of interest to declare. This work is part of the research project Greifswald Approach to Individualized Medicine (GANI_MED). The GANI_MED consortium was funded by the Federal Ministry of Education and Research and the Ministry of Cultural Affairs of the Federal State of Mecklenburg, West Pomerania (03IS2061A). We are grateful to Jette Anklam for technical assistance.
Executive summary
- Clinical proteomics has come a long way in terms of technology development, biochemical characterization and bioinformatics to identify molecular signal pattern and stratification of diseases.
- Human biofluids possess complex composition, high dynamic range and high variability.
- In-depth characterization of human biofluids is important for functional studies to discover biomarker candidates.
- Many human biofluids have not been comprehensively characterized yet; proteomic profiling of further specimens is far from exhaustive and further investigation is needed.
- Central database repositories of biofluid proteomes are needed.
- Only a few clinical applications have resulted from proteomic biofluid studies.
- First applications towards personalized proteomics have recently been performed and will hopefully lead towards clinical implementations.
Key terms
Biofluids: Human liquid to be secreted, excreted or released from healthy or pathological tissue and containing a high proportion of body water.
Dynamic range of proteins: Within human biofluids this requires sophisticated sample preparation, such as (i) depletion of high abundant proteins, (ii) extensive pre-fractionation on peptide or protein level, (iii) targeted protein analysis by immunoaffinity or (iv) enrichment of peptide classes (glycopeptides or N-terminal peptides).
Human biofluid diversity: All in-depth proteomic analyzed biofluids so far consist of several thousands of proteins, whereas about 4 to 44% are unique proteins to the appropriate biofluid.
Population and phenotype-based study cohorts: Allow age and gender matched phenotypic characteristic data analysis. In addition, large scale proteomics is a promising approach towards personalized proteomics that aims to establish approved and accepted protein biomarkers in routine clinical analysis.
References
- Jehmlich N, Dinh KH, Gesell-Salazar M et al. Quantitative analysis of the intra- and inter-subject variability of the whole salivary proteome. J. Periodontal Res. 48(3), 392–403 (2013).
- Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1(11), 845–867 (2002).
- Veenstra TD, Conrads TP, Hood BL, Avellino AM, Ellenbogen RG, Morrison RS. Biomarkers: mining the biofluid proteome. Mol. Cell. Proteomics 4(4), 409–418 (2005).
- Anderson NL. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin. Chem. 56(2), 177–185 (2010).
- Koutroukides TA, Guest PC, Leweke FM et al. Characterization of the human serum depletome by label-free shotgun proteomics. J. Sep. Sci. 34(13), 1621–1626 (2011).
- Razavi M, Johnson LD, Lum JJ, Kruppa G, Anderson NL, Pearson TW. Quantification of a proteotypic peptide from protein C inhibitor by liquid chromatography-free SISCAPA-MALDI mass spectrometry: application to identification of recurrence of prostate cancer. Clin. Chem. 59(10), 1514–1522 (2013).
- Feig MA, Hammer E, Volker U, Jehmlich N. In-depth proteomic analysis of the human cerumen-a potential novel diagnostically relevant biofluid. J. Proteomics 83, 119–129 (2013).
- Farrah T, Deutsch EW, Omenn GS et al. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10(9), M110 006353 (2011).
- Kentsis A, Monigatti F, Dorff K, Campagne F, Bachur R, Steen H. Urine proteomics for profiling of human disease using high accuracy mass spectrometry. Proteomics Clin. Appl. 3(9), 1052–1061 (2009).
- Schutzer SE, Angel TE, Liu T et al. Distinct cerebrospinal fluid proteomes differentiate post-treatment lyme disease from chronic fatigue syndrome. PloS One 6(2), e17287 (2011).
- de Almeida Pdel V, Gregio AM, Machado MA, de Lima AA, Azevedo LR. Saliva composition and functions: a comprehensive review. J. Contemp. Dent. Pract. 9(3), 72–80 (2008).
- Zhang A, Sun H, Wang P, Wang X. Salivary proteomics in biomedical research. Clin. Chim. Acta. 415, 261–265 (2013).
- Bandhakavi S, Stone MD, Onsongo G, Van Riper SK, Griffin TJ. A dynamic range compression and three-dimensional peptide fractionation analysis platform expands proteome coverage and the diagnostic potential of whole saliva. J. Proteome Res. 8(12), 5590–5600 (2009).
- Boja ES, Rodriguez H. The path to clinical proteomics research: integration of proteomics, genomics, clinical laboratory and regulatory science. Korean J. Lab. Med. 31(2), 61–71 (2011).
- Mischak H, Allmaier G, Apweiler R et al. Recommendations for biomarker identification and qualification in clinical proteomics. Sci. Transl. Med. 2(46), 46ps42 (2010).
- Marx H, Lemeer S, Schliep JE et al. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 31(6), 557–564 (2013).
- Barbhuiya MA, Sahasrabuddhe NA, Pinto SM et al. Comprehensive proteomic analysis of human bile. Proteomics 11(23), 4443–4453 (2011).
- Rolland AD, Lavigne R, Dauly C et al. Identification of genital tract markers in the human seminal plasma using an integrative genomics approach. Hum. Reprod. 28(1), 199–209 (2013).
- Zegels G, Van Raemdonck GA, Tjalma WA, Van Ostade XW. Use of cervicovaginal fluid for the identification of biomarkers for pathologies of the female genital tract. Proteome Sci. 8, 63 (2010).
- Zhou L, Zhao SZ, Koh SK et al. In-depth analysis of the human tear proteome. J. Proteomics 75(13), 3877–3885 (2012).
- Higashi T. Salivary hormone measurement using LC/MS/MS: specific and patient-friendly tool for assessment of endocrine function. Biol. Pharm. Bull. 35(9), 1401–1408 (2012).
- Loo JA, Yan W, Ramachandran P, Wong DT. Comparative human salivary and plasma proteomes. J. Dent. Res. 89(10), 1016–1023 (2010).
- Gobezie R, Kho A, Krastins B et al. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 9(2), R36 (2007).
- Aregger F, Pilop C, Uehlinger DE et al. Urinary proteomics before and after extracorporeal circulation in patients with and without acute kidney injury. J. Thorac. Cardiovasc. Surg. 139(3), 692–700 (2010).
- Bondar OP, Barnidge DR, Klee EW, Davis BJ, Klee GG. LC-MS/MS quantification of Zn-alpha2 glycoprotein: a potential serum biomarker for prostate cancer. Clin. Chem. 53(4), 673–678 (2007).
- Dubois V, Delort L, Mishellany F et al. Zinc-alpha2-glycoprotein: a new biomarker of breast cancer? Anticancer Res. 30(7), 2919–2925 (2010).
- Fung ET. A recipe for proteomics diagnostic test development: the OVA1 test, from biomarker discovery to FDA clearance. Clin. Chem. 56(2), 327–329 (2010).
- Surinova S, Schiess R, Huttenhain R, Cerciello F, Wollscheid B, Aebersold R. On the development of plasma protein biomarkers. J. Proteome Res. 10(1), 5–16 (2011).
- Zhang Q, Fillmore TL, Schepmoes AA et al. Serum proteomics reveals systemic dysregulation of innate immunity in type 1 diabetes. J. Exp. Med. 210(1), 191–203 (2013).
- Johansson A, Enroth S, Palmblad M, Deelder AM, Bergquist J, Gyllensten U. Identification of genetic variants influencing the human plasma proteome. PNAS 110(12), 4673–4678 (2013).
- Chen R, Mias GI, Li-Pook-Than J et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148(6), 1293–1307 (2012).
- Lin D, Alborn WE, Slebos RJ, Liebler DC. Comparison of protein immunoprecipitation-multiple reaction monitoring with ELISA for assay of biomarker candidates in plasma. J. Proteome Res. 12(12) 5996–6003 (2013).