Big breakthrough in small science: new MS technology could reshape sample analysis
MS is a powerful analytical technique, but it suffers from a major drawback: approximately 99% of the sample is typically lost before analysis begins. A novel method created by a research team from Brown University (RI, USA) greatly reduces this loss, enabling a more accurate and efficient sample analysis.
The high rate of sample loss limits the potential of MS by reducing accuracy and sensitivity, wasting resources and complicating sample preparation, which can introduce further errors. To address these challenges, the team devised a new method for transferring ions analyzed by MS, termed a nanopore ion source, which significantly minimizes sample loss and preserves almost the entire sample.
“The conventional technique for producing ions for MS, called electrospray ionization, basically involves a very sharp needle getting placed just in front of the mass spectrometer, hitting it with an electric field that pulls out a spray of charged droplets that eventually dry out to produce bare ions that make it into the mass spectrometer from open air,” commented Nicholas Drachman, a Physics PhD student at Brown University who led the project.
“Basically, it’s a process where you’re really spraying your sample all over the place to produce these ions and only get a tiny portion of them into the mass spectrometer’s vacuum for analysis. Our approach skips all of that.”
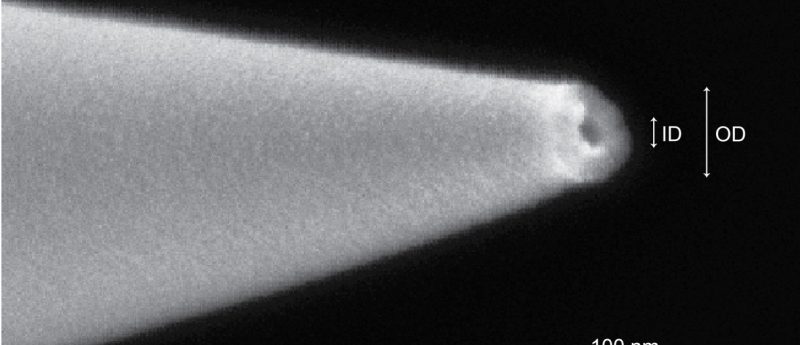
The crucial innovation is a tiny capillary developed by the researchers, with an opening ~30 nanometers wide. In contrast, the typical needle size used in electrospray has an opening of ~20 micrometers, which is approximately 600 times larger.
This new nanotube allows for the transfer of ions dissolved in water to be transported directly into the vacuum of a mass spectrometer, eliminating the need to create a spray of droplets that must be dried in order to access the ions. MS typically draws in a substantial amount of gas during the transfer of ions, however this new method prevents any gas from being drawn in, eliminating the need for any gas removal.
You may also be interested in:
- The new frontier: using LC–MS/MS for quantitative assays to support gene therapies
- Validation of Mitra® VAMS® as a blood collection technique for trace elements analysis using ICP-MS/MS
- The 2024 Summer Olympic Games: the evolving role of mass spectrometry in sports and athlete exposome analysis
The Brown team drew inspiration from nanopore sequencing in DNA and aims to market their technology for widespread use by protein researchers. This could achieve the long-standing goal of sequencing proteins one amino acid at a time.
“Proteomics have not seen the same advances as genomics in the last two decades, and so there’s been this this hunger for a technology that can improve analysis of proteins. By getting rid of that sample loss problem, it should enable these much more sensitive analyses to be possible, like sequencing the amino acids in a protein molecule one-by-one and in sequential order. This is the blue-sky idea that has motivated our work,” explained Derek Stein, a professor of Physics at Brown University and an author on the research paper.
Over the past 10 years the research team developed their method, starting with creating their own custom mass spectrometer that could contain the unique ion source in a vacuum. This was distinct from conventional designs where the ion source is positioned separate from the device and remains in open air.
The key component of their transfer device was built by using a specialized machine to heat a glass tube and then delicately pull it apart to create an extremely small opening at the tip, completely invisible to the naked eye. It was successfully demonstrated that performing ion analysis with their new transfer method matches detections conducted using traditional methods, but with far less sample loss.
The results of their study serve as proof of concept for the method. In the future, the researchers aim to unlock the full potential of their nanopore ion source.
“We need to show that this can improve the workflow of proteomic analyses,” Drachman stated. “We’d like to take that to the next level and make it something that will improve the science of researchers throughout the field.”